Here are some follow-up items related to two of my previous posts: “The FDA, the USPTO and the Pharmaceutical Industry” and “Why The US Healthcare Care System Costs So Much – Part Three – Overtreatment”
The FDA
First, the FDA, again. There were two articles published this week (August 15, 2017) illuminating more on how the FDA favors the drug and device industries.
In the first article, entitled, “Characteristics of Pre-approval and Post-approval Studies for Drugs Granted Accelerated Approval by the US Food and Drug Administration”; Huseyin Naci, PhD, MHS; Katelyn R. Smalley, BSc; Aaron S. Kesselheim, MD, JD, MPH; JAMA. 2017;318(7):626-636, the authors analyze the research studies for 22 drugs that were given 24 indications under the accelerated approval process of the FDA. Accelerated approval is applied to drugs that treat serious or life-threatening conditions (like cancer, HIV, etc.). “Pre-approval” means studies that were done before the drug was FDA approved and “Post-approval” means the drug is studied after it is released to the public after being approved by the FDA. The post-approval time is when drug companies sell their drugs to the medical community and MAKE MONEY!
Remember that the lower the number of people in studies, the more unreliable the studies are.
There were some significant concerns voiced in this article which should make you take pause about the FDA’s behavior:
All of the research studies used “surrogate” measures to “prove” the drug is effective. A surrogate measure is an indirect measure that doesn’t prove you, the patient, will actually benefit from the drug. Here are some surrogate measures compared to a true clinical outcome measure:
| Surrogate measure | True Clinical Outcome measure | Why is this a problem? |
| Progression Free Survival (the amount of time a cancer tumor doesn’t grow and you are still alive) | Survival (you are still alive) | You can have a non-progressing tumor that suddenly activates and kills you in the same amount of time as if you had no treatment. You’re dead within the same amount of time with or without the drug |
| Increase in mammography rate | Breast Cancer Survival | Improving a process to increase a “process measure,” like a rate, doesn’t necessarily mean a patient will actually benefit from the intervention, in this case, not die. |
Here are some excerpts from this article:
“The clinical trial evidence for therapeutic agents granted accelerated approval by the FDA between 2009 and 2013 shows that 14 of 24 indications for these drugs entered the market on the basis of single intervention-group studies that enrolled a median of 132 patients, which some investigators would consider a small number.”
“Half of required confirmatory studies were completed a minimum of 3 years after the approved drug was on the market.” [Meaning, the drug was on the market for a long time before the drug company completed a confirmatory study. During that time, people could have been harmed and gotten no real clinical outcome improvement. But, the drug company MAKES MONEY!]
“The quality and quantity of post-marketing studies required by the FDA to confirm clinical benefit varied widely across indications. There were few statistically detectable differences in the key design features of trials conducted before and after approval. Nonrandomized studies were common in the accelerated approval pathway both before (60%) and after (44%) market entry. Even though the majority of completed studies showed positive results in the post-marketing period, all completed confirmatory studies demonstrating drug benefit evaluated surrogate measures of disease activity rather than clinical outcomes”.
“For the 10 accelerated approvals between 2009 and 2013 that have since had their requirements fulfilled and labels updated, all of which were for cancer indications, the studies used to confirm clinical benefit tested surrogate measures. The FDA’s senior scientists consider overall survival to be the most dependable end point in clinical trials of cancer drugs. Yet overall survival was among the pre-specified primary end points in only 5% of required confirmatory studies.” [In other words, true effectiveness has never been proven.]
“Another finding from the current study is the slow progression of some post-approval studies. A recent Government Accountability Office report criticized the FDA’s oversight of drugs approved on the basis of surrogate measures.” [?Collusion between the FDA and drug companies?]
“For 14 (58%) of 24 indications granted accelerated approval from 2009 to 2013, results from required confirmatory studies were not available after a median of 5 years of follow-up, and (42%) of 19 incomplete confirmatory studies were either terminated or delayed by more than 1 year. [And the drug companies were making tons of money without confirming effectiveness.]
“Confirmatory studies failed to demonstrate clinical benefit in 2 indications granted accelerated approval between 2009 and 2013. According to the Code of Federal Regulations, the FDA may withdraw a therapeutic agent if confirmatory studies fail to verify its clinical benefit. However, according to publicly available documents, the FDA has neither rescinded its approval nor imposed additional requirements for these 2 indications. Historically, the FDA has rarely withdrawn an indication during the 25 years since the accelerated approval pathway was established. [Only one drug in 25 years. The drug didn’t improve outcomes and increased toxicity. ]
Makes you wonder, doesn’t it?
Here’s the second article: “Characteristics of Clinical Studies Used for US Food and Drug Administration Approval of High-Risk Medical Device Supplements”; Sarah Y. Zheng, MD; Sanket S. Dhruva, MD, MHS; Rita F. Redberg, MD, MSc: JAMA. 2017;318(7):619-625. Rather than going into the detail as I did in the above article, I’ll just give you the summary (“cliff notes”) version printed within the article:
“Question: What is the quality of clinical studies and data used to approve modifications to high-risk devices by the US Food and Drug Administration (FDA) panel-track supplement pathway?
Findings: In this descriptive study of 83 clinical studies for 78 panel-track supplements approved between 2006 and 2015, 45% were randomized clinical trials and 30% were blinded. Of the 150 primary end points in these studies, 81%were surrogates and 38% were compared with controls.
Meaning: There are limitations in the quality of the studies and data evaluated by the FDA to support modifications of high-risk devices.
CONCLUSIONS AND RELEVANCE: Among clinical studies used to support FDA approval of high-risk medical device modifications, fewer than half were randomized, blinded, or controlled, and most primary outcomes were based on surrogate end points. These findings suggest that the quality of studies and data evaluated to support approval by the FDA of modifications of high-risk devices should be improved.”
So, for drugs for life-threatening illnesses with serious side-effects and toxicities, and for medical devices that are considered high-risk, the FDA approves based on data that is, at most, only suggestive. And when a post-approval study demonstrates no effectiveness, only once is 25 years, has the FDA rescinded its approval.
Things that make you go “Hmmmm”, right?
Overuse (Overtreatment)
Here are two articles that show that physicians and APCs are doing things they shouldn’t, costing million of dollars and with risks to patients as part of the clinical scenario. And, the recommendations and conclusions in the articles are examples of the legacy way of thinking within the medical community,.
The first article is: “Eliminating Creatine Kinase–Myocardial Band Testing in Suspected Acute Coronary Syndrome”; Matthew D. Alvin, MD, MBA,MS, MA; Allan S. Jaffe, MD; Roy C. Ziegelstein, MD,MACP; Jeffrey C. Trost, MD; JAMA Internal Medicine Published online August 14, 2017.
In this article, the authors describe all of the research that proves that doing a “creatine-kinase-myocardial-band test” (CK-MB test) has no value in diagnosing a heart attack because, for the last 15-20 years, a better test, called the troponin test, has been proven to be more sensitive and specific for a heart attack. Despite this, providers are still ordering CK-MB tests, costing millions of dollars a year for a test with “no value”. Here’s just one short excerpt from the article:
“Once the cornerstone of AMI diagnosis, CK-MB has not yet been eliminated from practice despite considerable evidence supporting cTn (cTroponin) as the preferred biomarker. Data published after distribution of the ACC/ESC/AHA3-5 recommendations show the these clinical practice guidelines have not succeeded in refining practice. Specifically, CK-MB is still used in many US clinical pathology laboratories and US EDs”
The article goes on to say that there are clinical situations that have been cited where doing a CK-MB has actually negatively impacted patient care.
In a non-health care environment, like manufacturing, the industry would simply eliminate the opportunity to “do the wrong thing”. This would be the highest ranking and most effective intervention for mistake proofing the “diagnosis of heart attack” process. Remember this mistake proofing “solution starter”?
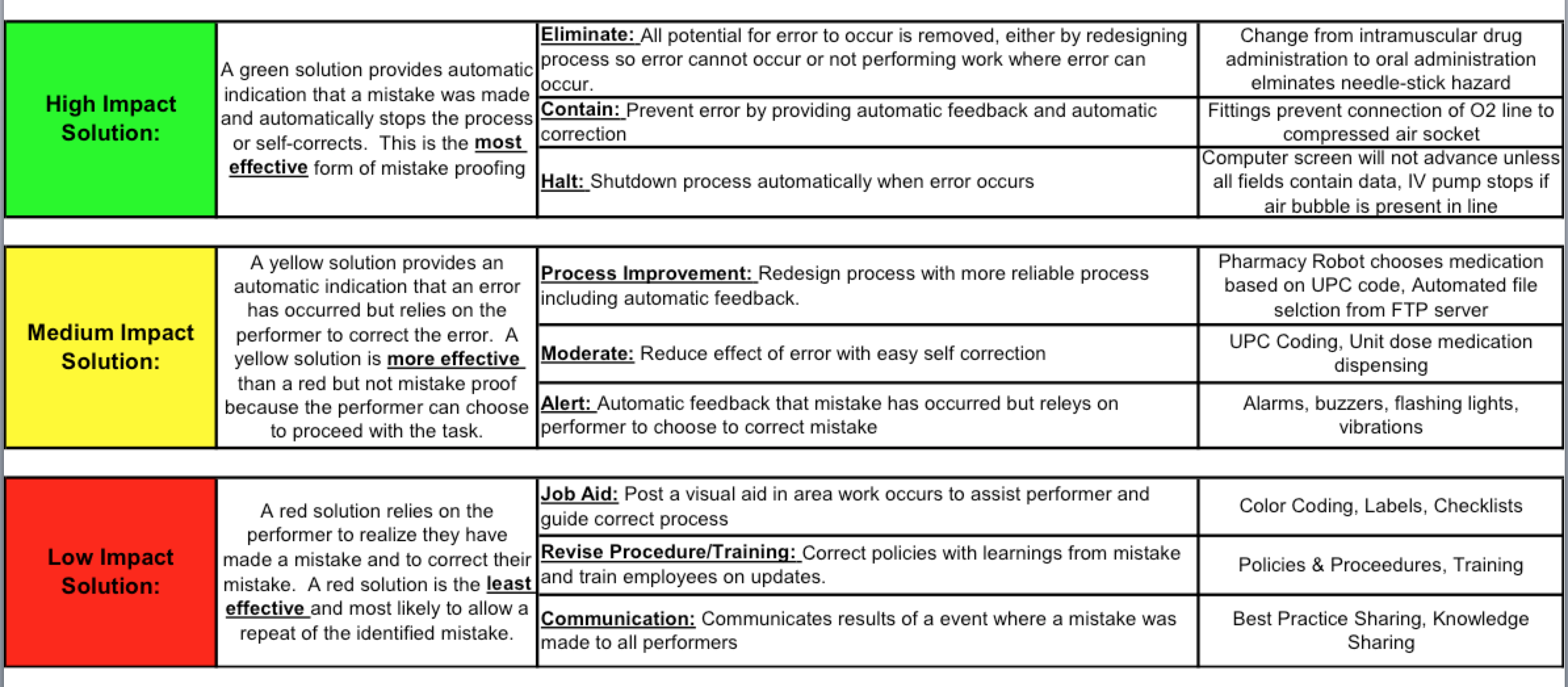
The highest impact solution is eliminating the step or option that causes the error. In a true “error reduction” environment, you would just eliminate the option of ordering a CK-MB for a heart attack. It wouldn’t be on any lab order sheet. If a physician ordered it, it would be rejected if the indication was “rule out heart attack”.
But, this rarely occurs with health care because they mostly use the low impact mistake proofing solutions (the ones that are RED!) The above article cites another article by Larochelle et al that implemented the following:
1. Create an institutional specific guideline (when there already are national guidelines by the ACC and AHA) (Revise Procedure/Training, second weakest impact intervention)
2. Educational sessions with practitioners (Revise Procedure/Training, second weakest impact intervention)
3. Dissemination of quick reference cards with the guideline elements on it (Job Aid, third weakest impact intervention)
4. Removal of the CK-MB from the order sets in the electronic medical record. (Eliminate, the highest impact intervention)
The result was, there were zero CK-MB ordered during the 12th month after the changes were made. They didn’t assess the effectiveness of the individual elements described above, but, I would guess that the fourth item was the truly effective solution as it ELIMINATED the option to order the test. The other three are red-zone solutions. The authors actually cite:
“Multiple other groups, including Mayo Clinic, simply removed CK-MB from routine order sets and found 80.0% to 99.8% reductions in CK-MB orders with significant cost savings and without negative impact on patient care or missed AMI diagnoses”
But, then, the authors recommend the following (non-italicized comments are mine):
The methodology and theory of the blueprint and quality improvement initiative are based on the US Health Resources and Services Administration strategies for developing and implementing a quality improvement initiative, with a focus on education, action, and measurement of results. The leader of each institution should assess applicability of this blueprint based on the CK-MB ordering frequency at their respective institution.
1. Design and implement a hospital-wide educational campaign. [Red Zone Solution. This whole following section is totally unnecessary.]
a.Prior to implementation, it is important for health care leaders to establish sufficient organizational readiness for change, specifically in conjunction with these stakeholders, whose ordering practices will be affected by such changes. Tools to measure readiness for change have been previously studied and may be used.
b. Collaborate with physician representatives from the departments of cardiology, internal medicine, and emergency medicine. Frontline physicians in these departments order the majority of cardiac biomarkers within most health care institutions and are the primary stakeholders in the current system of care for ACS patients.
c. Academic institutions must engage the house staff as members of the quality improvement team for an effective initiative.
d. Collaborate with secondary stakeholders, such as pathology and/or laboratory staff, to acquire insight and advice on these changes.
c. Inform physicians that CK-MB adds to the health care system financial burden without adding value to patient care.
d. Present the evidence supporting elimination of CK-MB and exclusive use of cTn to diagnose AMI, identify reinfarction, and estimate infarct size. Education may be provided through various venues, including lectures, pocket cards (Figure 2), online modules, social media demonstrations, and simulations.
2. Partner with information technology and/or laboratory medicine staff to remove CK-MB from standardized ACS routine order sets. Doing this simple step alone has been shown to significantly reduce CK-MB ordering (Table). [Green Zone Solution. Elimination is the highest impact. This is all that is needed.]
3. Partner with information technology and/or laboratory medicine staff to create and integrate a best practice alert (BPA) into the CPOE to appear when clinicians order CK-MB, such as:
“According to national guidelines, troponin is the preferred biomarker for detecting myocardial injury; CK-MB is only appropriate if troponin testing is unavailable.” [Red Zone Solution.]
4. Measure data pre-intervention and post-intervention (efficacy points). [This is necessary. Measurement System to document if successful or not]
a. Number of cTn and CK-MB tests ordered, including stratification by department and patient setting (ED, inpatient, medicine vs non medicine units, ICU vs non-ICU).
b. Incidence, missed diagnoses ,and mortality of AMI to ensure patient safety.
c. Review cases where CK-MB is still ordered to determine if it provides value.
d. If necessary, track usage by physician to develop performance feedback profiles.
e. Calculate reduction in charges to patients and health plans, as well as any decrease in hospital costs.
Because health care is mostly a consensus environment rather than a just do it environment, the above has a huge amount of unnecessary work (anything in the red zone solution area) and lead time to implementation delays because the one green zone intervention is an ELIMINATION solution. The only work that really needs to be done is inform the practitioners that current national guidelines eliminated the ordering of the CK-MB test for heart attacks and it will no longer be available for that indication. If an institution wants to insert an alert that pops up in an EMR to inform the practitioner, that should be the only additional work. All of the rest of the above is WASTE, and health care systems already have too much waste already.
The problem is no one in health care has the balls to just say NO when something is inappropriate! You have to “baby” the practitioners through it even when it is extremely obvious. I’m all for Prochaska’s readiness to change and getting buy-in and engagement for many things (like actual process re-design and implementation) but not in a case like this where it is cut and dried as to what should be done and the intervention is a single step (really, elimination of a step). In the Lean world, they would call a lot of the red zone interventions above “over-processing” (doing more work than necessary to get the desired result).
The second article in this section is: “Unnecessary Staging Imaging in Early-Stage Breast Cancer”; Heather R.Wolfe, MD; Arjun Gupta, MD; Navid Sadeghi, MD; JAMA Internal Medicine Published online August 14, 2017. In this article, the authors go through the literature and proclaim:
“Patients with early disease confined to the breast with no or limited lymph node involvement require no further staging imaging.”
Then they go on to say:
“Although breast cancer care is now largely multidisciplinary, it is often first suspected or diagnosed by primary care physicians. They may be unaware of the needlessness of routine staging imaging in otherwise asymptomatic patients.”
This statement is not supported by any data in the article. It is just blaming! I personally don’t know any Primary Care Physicians who would order the battery of imaging tests that would constitute staging for breast cancer. Stating this in the article is completely inappropriate.
Finally, the authors bemoan the overuse, but don’t put forth one single recommendation as to how to reduce or stop unnecessary staging in the patients with early disease confined to the breast with no or limited lymph node involvement. This seems to me to be another “eliminate” [Green Zone] candidate. What should happen is, any imaging center that gets a request for breast cancer staging has to demand the clinical information that supports the ordering of the testing. If the clinical information is “the patient has early disease confined to the breast with no or limited lymph node involvement”, then the testing should be cancelled. ELIMINATE the testing.
I actually approached a large radiology group in one state where I was working and asked if they would do this.
Radiology request forms usually have three parts: 1) the test you want to order (like, X-ray of the leg), 2) why you want the test (like, “suspect a fracture”), and 3) some clinical information (like, “patient fell 30 feet from a scaffolding. Leg pain. Leg is deformed on examination.”).
Nowadays, it is hard for a PCP, Hospitalist or ED practitioner to remember and keep track of which radiology test is best for the clinical situation at hand, especially tests like CT-scans, MRI’s and Ultrasounds. But, Radiologists should know, shouldn’t they? Well, THEY DO! For example, their specialty society publishes the indications for all of these high tech tests that are used in breast cancer staging.
So, I asked them if, when they got a request for an imaging study and the request form information didn’t match the indications for the test that was ordered, would they contact the ordering practitioner and either get further information that would justify the test or tell the ordering practitioner they had ordered the wrong test and an alternative test was the best test and they would like to substitute the best test.
This sounds simple, cost effective and helpful for the patient (because, when the wrong test is done, eventually the right test needs to be done. For x-ray tests, patients get extra radiation exposure. They also might be subject to increased out of pocket costs because they needed two tests instead on one).
What do you think the Radiologists said? You would think it was, “Sure this sounds great, and we are well-trained and can do this! After all, the guidelines for which tests should be done in clinical situations are our Radiological Society Guidelines. We HAVE to know them to be board certified!”
Nope. Instead it was , “Oh no. Our role is to do whatever the ordering practitioner orders.” (Does this sound like Nazi Germany to you? “I’m just following orders!”).
So, I said, “You know, when I have a patient in my exam room with abdominal pain and I think they have appendicitis, I don’t send them to the surgeon ordering an appendectomy and he/she just does the appendectomy. I send them to the surgeon and ask for a surgical evaluation and a recommendation as to the next course of action. Maybe that’s an appendectomy and maybe it’s not. That’s what specialists do, other than you. Are you saying you don’t want to behave like a specialist?
Their answer to this was, “We aren’t going to do that. We’ll do whatever the ordering practitioner orders. If you want better compliance by ordering practitioners with the radiological guidelines, you’ll have to figure out how to do that.”
And so there it is for imaging studies. Radiologists don’t want to act like specialists who know what the best thing to do is according to their own society’s guidelines!” Want to know why? BECAUSE THEY MAKE MORE MONEY DOING THE EXTRA, WRONG TESTS!!!
After all, it’s all about the money.